|
|
|
|
|
| An evaluation of ChemNMR, the NMR prediction software within ChemDraw Ultra |
|
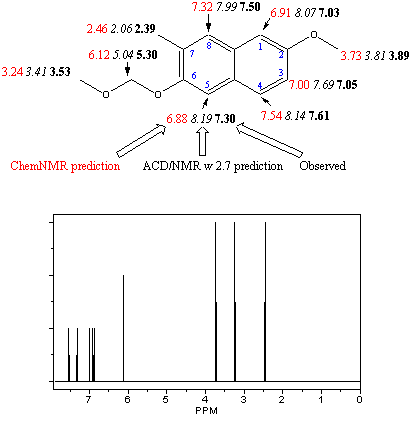
By Tony Herlt, Research School of Chemistry, Australian National University, Canberra, ACT 020, Australia I recently received a copy of ChemDraw Ultra version 5.0 for Windows, and was especially interested in checking out its NMR predicting features. I already had experience using Advanced Chemistry Development's web based NMR predictor ACD/NMR w 2.7 , and wanted to compare how the two programs predicted chemical shifts for a reasonably complicated molecule where all resonances had been assigned. What is ChemNMR? ChemNMR was developed by Upstream Solutions to be run within ChemDraw Ultra. The program is invoked by first selecting a chemical structure that one wants predicted, then going to the <Estimate> menu and selecting <1H NMR shifts> or <13C NMR shifts>. Calculation is very rapid and estimated chemical shifts are displayed adjacent to the relevant nuclei. Chemical shifts are also displayed as a line spectrum, and a more detailed account of the estimation is provided as a text file in Notepad. The program works by breaking the structure down into one or more substructures that provide the base value for the estimated shift. Additivity rules are then applied and the base value is incremented to provide the final value. In the case of 1H NMR, the shifts are predicted for a non-polar solvent. Hydrogens bonded to hetero atoms e.g. OH, NH, and SH are not calculated. Proton-proton coupling constants are also not predicted. Elements of the first four rows and a few from the fifth row of the periodic table are recognized. Using ChemNMR to predict 1H NMR shifts for one of my compounds I am currently working on substituted napthalenes and applied the program to a compound I recently synthesized and fully assigned the 1H NMR spectrum of. Figure 1 shows the result for 2-methoxy-6-(methoxymethoxy)-7-methylnapthalene. For each hydrogen in the compound there are 3 values for the chemical shift. The first is the value predicted by ChemNMR; the second (in italics) is that predicted by ACD/NMR w 2.7 and the third (in bold) is the observed chemical shift in CDCl3. ChemNMR was more accurate in it. s prediction than ACD/NMR w 2.7 for the CH3 group on the aromatic ring, but did not fare so well with the other aliphatic protons, in particular the CH2 of the MOM group. However with the aromatic protons Chem NMR was clearly superior in its prediction. In the next synthetic step an isobutyryl group is introduce at C-5 which now makes the protons in the CH2 of the MOM group and the CH3's of the isobutyryl group non-equivalent. Neither program predicted this non-equivalence.
Figure 1: Predicted and Observed 1H NMR Shifts for 2-methoxy-6-(methoxymethoxy)-7-methylnapthalene Using ChemNMR to predict 1H and 13C NMR shifts for a compound from the chemical literature In selecting a suitable compound my criteria were that all chemical shifts for hydrogen and carbon had been assigned and there were as many different CHx groups as possible to provide an objective evaluation. I decided on a couple of natural products called secoiridoids that had been isolated from the European olive and fully characterized ( P. Gariboldi, G. Jommi and L. Verotta, Phytochemistry. 1986, 25, 865). The two compounds are diastereoisomeric about C-6 and both their 1H and 13C NMR spectra were run in CDCl3. Figure 2 shows the predicted 1H NMR for the two compounds with the order within each series of values the same as in Figure 1. In the aromatic region ChemNMR was clearly superior to ACD/NMR w 2.7 in its predictions. This was also evident in the benzylic protons, the two CH3 groups and all the protons in the dihydropyran ring except H-4. Neither program predicted non-equivalence for the allylic protons although again ChemNMR was closer to the observed chemical shifts. ACD/NMR w 2.7 did predict non-equivalence for the protons on the carbon between the dihydropyran ring and the carbonyl, and was more accurate in its prediction for one of the structures. Both programs predicted the same shifts for the two structures, indicating that there is still a long way to go before subtle NMR spectral changes arising from stereochemical differences can be predicted by software. |
|

Figure 2: Predicted and Observed 1H NMRShifts for the Secoiridoids |
|
Figure 3 shows the 13 C NMR predictions for the same two compounds. Here both programs performed well with the exception of C-4 of the dihydropyran ring. Neither program again recognized the stereo-chemical difference between the two structures.
|

Figure 3: Predicted and Observed 13C NMR Shifts for the Secoiridoids |
|
So is there a winner?
I would have to say that for 1H NMR, based on the above results and predictions for many other compounds that ChemNMR is superior to ACD/NMR w 2.7 for accuracy. For 13C NMR there is little difference, both programs do a good job and when there are significant differences between predicted and observed shifts, both programs tend to be in error. One must bear in mind however that this comparison was done only with the web based NMR predictor from Advanced Chemistry Development. Their program is more powerful in that it will predict coupling constants, and spectra can be simulated at different spectrometer frequencies and at different line widths. I do not have access to their stand alone PC version which may predict differently. A comparison between this version and ChemNMR would be interesting. How I rate the software On a scale of 1 to 5 I would give ChemNMR a 4. For ChemDraw Ultra as a whole I would also give a 4. One of the tools that take some of the tedium out of chemistry is <analyze structure> which calculates molecular weight and elemental analysis from a structure. The other is <Convert structure to name> which uses the Beilstein AutoNom system to derive IUPAC names from a structure (unfortunately it does not recognize stereochemistry, unlike Chem 4-D Draw pro from ChemInnovation software which does). When I started my chemical career over a quarter of a century ago I used to draw structures with a ruler and a stencil. We have come a long way since then with chemistry drawing programs and their add-ons. I wonder what they will be like a quarter of a century from now. Tony Herlt, 15th March 1999
|

|
ChemNMR is bundled with particular variants of ChemDraw and ChemOffice which can be purchased directly from CambridgeSoft. |
|
|
|
| [News] [Sitemap] | [Us] |
|
|
|